
We serve Chemical Name:2-Chloro-4-fluoro-5-nitrotoluene CAS:112108-73-3 to global customers since 2007, Pls send inquiry to info@nbinno.com or visit www.nbinno.com our official website should you have any interests. This site is for information only.
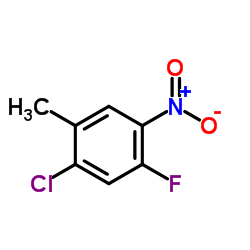
Chemical Name:2-Chloro-4-fluoro-5-nitrotoluene
CAS.NO:112108-73-3
Synonyms:1-Chloro-5-fluoro-2-methyl-4-nitrobenzene;Benzene, 1-chloro-5-fluoro-2-methyl-4-nitro-;1-Chloro-5-Fluoro-2-Methyl-4-Nitro-Benzene;2-Chloro-4-fluoro-5-nitrotoluene;WNR DG BF E1
Molecular Formula:C7H5ClFNO2
Molecular Weight:189.572
HS Code:2904909090
Physical and Chemical Properties:
Melting point:34-36ºC
Boiling point:267.6±35.0 °C at 760 mmHg
Density:1.4±0.1 g/cm3
Index of Refraction:1.549
PSA:45.82000
Exact Mass:188.999283
LogP:2.36
Material Safety Information (Applicable for Hazard Chemicals)
RIDADR:
Packing Group:
Contact us for information like 1-Chloro-5-fluoro-2-methyl-4-nitrobenzene chemical properties,Structure,melting point,boiling point,density,molecular formula,molecular weight,WNR DG BF E1 physical properties,toxicity information,customs codes,safety, risk, hazard and MSDS, CAS,cas number,1-Chloro-5-Fluoro-2-Methyl-4-Nitro-Benzene Use and application,1-Chloro-5-fluoro-2-methyl-4-nitrobenzene technical grade,usp/ep/jp grade.
Related News: The Japanese drugmaker is injecting $126 million into its Thousand Oaks facility to boost manufacturing and support new product lines, Pacific Coast Business Times first reported. Takeda was joined by the city’s mayor, Claudia Bill-de la Peña, for a groundbreaking event on Thursday, July 22. N-BOC-Diethanolamine manufacturers Takeda decided to expand the collaboration given the great progress” the two companies have made since initiating their joint research last December, said Sarah Sheikh, head of Takeda’s neuroscience therapeutic unit, in a statement to Fierce Biotech.
Neuroscience drug development is hindered by the challenge of getting therapeutics into the brain and other parts of the nervous system, especially for large molecule treatments that struggle to cross the blood-brain barrier, Sheikh said.
“Large molecule therapeutics are the latest treatments to be developed for neurodegenerative diseases and technologies that improve their ability to cross the blood-brain barrier will help to maximize their benefit,” Sheikh said.
In steps PeptiDream with its TfR1 binding technology, which Sheikh said has the potential to address the blood-brain barrier penetration issue.
Sheikh declined to disclose specific targets of the pact but said the two companies have “several different programs” that they hope to bring into the clinic.
The Japanese Big Pharma is betting big on neuroscience after offloading seven programs in a deal worth up to $2 billion last June with Neurocrine Biosciences.
Takeda handed over two phase 2 assets as part of that pact, saying goodbye to candidates across schizophrenia, treatment-resistant depression and anhedonia, or the inability to feel pleasure.
Takeda’s neuroscience pipeline is farther behind than areas it’s more familiar with, including oncology, gastroenterology and rare genetics and hematology.
The company did reach a milestone in the space Wednesday, though, with a breakthrough therapy designation from the FDA for its phase 2 oral orexin agonist being studied in the chronic neurological disorder narcolepsy type 1.
The Big Pharma expects its first drug approval in neuroscience to be in fiscal year 2023, with a potential first-in-class therapy for Dravet Syndrome and Lennox-Gastaut Syndrome.
In March, Takeda regained the rights to that program from Ovid Therapeutics for a $196 million upfront payment and up to $856 million total. At the time, Takeda said it would initiate phase 3 studies in children and adults with the epilepsy syndromes in the second quarter of this year.
Also that month, Takeda said it would potentially pay more than $500 million for a five-program collaboration with BridGene, including hard-to-drug neurodegenerative disease targets.
Antibodies triggered by Sinovac Biotech’s (SVA.O) COVID-19 vaccine declined below a key threshold from around six months after a second dose for most recipients, but a third shot had a strong booster effect, a lab study showed.
Chinese researchers reported the findings from a study of blood samples from healthy adults aged between 18-59 in a paper published on Sunday, which has not been peer reviewed. https://bit.ly/3zGsxQt
Among participants who received two doses, two or four weeks apart, only 16.9% and 35.2% respectively still had neutralising antibodies above what researchers regard as a detectable threshold level six months after the second shot, the paper said.
Those readings were based on data from two cohorts involving more than 50 participants each, while the study gave third doses of the vaccine or placebo to a total of 540 participants.
Researchers said it was unclear how the decrease in antibodies would affect the shot’s effectiveness, since scientists have yet to figure out precisely the threshold of antibody levels for a vaccine to be able to prevent the disease.
Apart from durable antibodies, other components in humans immune systems such as T cells and B cell memory elicited by the vaccine may also contribute to protection, researchers involved in the study said, although the study did not provide data on those factors.
“In the short-to-medium term, ensuring more people complete the current two-dose schedule of CoronaVac (Sinovac’s vaccine) should be the priority,” the paper said.
Participants in some cohorts who received a third dose of the Sinovac shot about six months after the second showed around a 3-5 fold increase in antibody levels after a further 28 days, compared with the levels seen four weeks after the second shot.
As of end-June, Sinovac has delivered more than 1 billion doses of the vaccine, a major vaccination tool in China, Brazil, Indonesia and Chile.
Other COVID-19 vaccine makers are seeking to get authorisation for booster shots. Pfizer (PFE.N) and partner BioNTech (22UAy.DE) said earlier this month they had planned to ask U.S. and European regulators within weeks to authorise a booster dose of its COVID-19 vaccine, based on evidence of greater risk of infection six months after inoculation and the spread of the highly contagious Delta variant. read more
Siti Nadia Tarmizi, a senior health ministry official in Indonesia, told Reuters on Tuesday that clinical data from the country showed the reduced antibody level is still enough to provide protection, without providing details. read more
She said the immunisation advisory board recommends a booster vaccination 12 months after the second dose.
Indonesia has started giving booster shots produced by Moderna (MRNA.O) to medical workers this month, including those who had received Sinovac doses.
Philippines’ Health Secretary Francisco Duque said on Tuesday there is no recommendation yet from the country’s vaccine expert panel on giving a booster shot but experts are discussing the possibility.
Turkey has already started offering a third dose from either Sinovac or Pfizer to some people who have got Sinovac shots.
The research paper on Sinovac’s vaccine said that the study did not test the antibodies’ effect against more transmissible variants, and that further research was needed to assess antibody duration after a third shot.
The study was conducted by researchers at disease control authorities in Jiangsu province, Sinovac, and other Chinese institutions.
Drug manufacturer Emergent BioSolutions Inc (EBS.N) said on Friday it has received inquiries and subpoenas from a swathe of U.S. authorities related to its abilities to manufacture COVID-19 bulk drug substance.
Emergent came under regulatory scrutiny after an error led to millions of vaccine doses being ruined at its manufacturing facility in Baltimore, which was producing bulk drug substance for Johnson & Johnson’s (JNJ.N) COVID-19 vaccine.
The U.S. Food and Drug Administration (FDA) in April halted operations at the plant after it discovered that ingredients from AstraZeneca’s (AZN.L) COVID-19 vaccine, also being produced there at that time, contaminated a batch of J&J’s vaccines.
Emergent in a filing on Friday said it has received “preliminary inquiries and subpoenas to produce documents” from the lawmakers, the Department of Justice, the Securities and Exchange Commission, the Financial Industry Regulatory Authority, and Maryland and New York Attorneys General.
The Department of Justice declined to comment on the matter.
Emergent said it was producing and has produced documents as required in response and will continue to cooperate with the government inquiries.
An FDA inspection in April turned up a long list of sanitary problems and bad manufacturing practices at the facility, including failure to train personnel to avoid cross contamination of COVID-19 vaccines from J&J and AstraZeneca. read more
Emergent said on Wednesday production at the Baltimore facility will now resume, following remedial actions, additional reviews and collaboration with the FDA and its manufacturing partners. read more
An FDA spokesperson said on Thursday the facility has not yet received any authorization for a COVID-19 vaccine, but may produce vaccine substance in advance of future inspections by the health regulator.
BeiGene’s Brukinsa is already approved in the small indication of mantle cell lymphoma. With a new trial win, the drug is on track to open up a much larger market.
Brukinsa outperformed the combination of bendamustine and rituximab at staving off cancer progression or death in newly diagnosed patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL), BeiGene said Thursday.
Although detailed data from the phase 3 Sequoia trial remain under wraps, BeiGene characterized Brukinsa’s advantage as “highly statistically significant.” The win was for patients whose tumor didn’t have a resistance risk factor called del(17p).
The latest win adds onto Brukinsa’s recent success in previously treated CLL/SLL and puts it on a collision course with the other two BTK inhibitors in the key CLL battleground.
In a head-to-head phase 3 trial dubbed Alpine, Brukinsa showed it could trigger a significantly better response than AbbVie and Johnson & Johnson’s market-leading Imbruvica in patients with relapsed or refractory CLL/SLL.
Plus, in an important safety win, patients who received BeiGene’s drug were less likely to develop atrial fibrillation, or irregular heartbeat.
Now, with pivotal data available from two trials, BeiGene could seek FDA nods for both front-line and second-line CLL simultaneously.
When BeiGene unveiled the second-line win at the European Hematology Association’s virtual congress last month, Jane Huang, M.D., chief medical officer of hematology, told Fierce Pharma that “there may be opportunities” to file both sets of data in one FDA application.
The new data could support an approval and a favorable label, “thus positioning Brukinsa as a key BTK inhibitor in the CLL space, particularly if the asset continues to demonstrate differentiation that could translate into a commercial advantage,” SVB Leerink analyst Andrew Berens wrote in a Friday note.
As the first BTK inhibitor to hit the scene, Imbruvica owns the lion’s share of the market. In the first half of 2021, J&J collected $2.24 billion from Imbruvica global sales, while AbbVie reported $2.65 billion, including $551 million from profit sharing with J&J.
Meanwhile, AstraZeneca’s Calquence is catching up quickly with a first-half haul of $490 million thanks to 150% year-over-year growth. The majority, $445 million, came from the U.S.
Calquence’s growth suggests that Imbruvica’s first-mover advantage in CLL isn’t insurmountable, Berens said, adding that Brukinsa seems to have an even better profile than Calquence.
Still, Brukinsa is BeiGene’s first commercial drug in the U.S. Facing those Big Pharma companies with entrenched oncology presence won’t be cakewalk for the Chinese company.
Brukinsa’s first-quarter sales came in at $22.1 million, mostly from China, where the drug has been allowed for relapsed or refractory CLL/SLL since last June.
Apparently, a licensing agreement with Takeda worth a potential $3.5 billion wasn’t enough for PeptiDream this week. The biopharmaceutical company, which has a peptide-based drug discovery platform, has now inked a similar deal with Alnylam worth a potential $2.2 billion.
Alnylam, which focuses on RNAi therapeutics, has tapped PeptiDream to develop peptide-small interfering RNA (siRNA) conjugates that will help deliver RNAi therapeutics to tissues outside the liver.
PeptiDream will receive an undisclosed upfront payment from Alnylam, R&D funding over the term of the collaboration and may be eligible to receive up to $2.2 billion in biobucks.
Additional royalty payments are part of the deal for any Alnylam products that result from the collaboration as well.That’s $5.8 billion in promise for just one week, though biotech veterans will know that the chances of every cent of that being picked up remains historically low, and comes with many, many caveats in a sector built on high-risk, high-reward.
Still, that’s a great week for PeptiDream, and further validates its platform.
“Having solved delivery of RNAi therapeutics to the liver and made substantial preclinical progress on in vivo delivery to the central nervous system, eye and lung, we are now adding to our suite of delivery technologies that have the potential to enable efficient delivery of siRNA to even broader tissue types throughout the human body,” said Alnylam’s Chief Scientific Officer Kevin Fitzgerald, Ph.D.
The collaboration will see Alnylam choose receptors for PeptiDream’s discovery platform, which will then select peptides to match.
Alnylam will then generate siRNA conjugates and perform studies to select the final peptides to move forward. The companies could develop multiple treatments from the partnership.
The companies did not disclose the exact diseases they will be working on, beyond the aim to deliver treatments to tissues outside the liver. Alnylam has four strategic therapeutic areas: genetic medicines, cardio-metabolic diseases, infectious diseases and central nervous system/ocular diseases.
This is the latest collaboration for PeptiDream that could be worth multiple billions of dollars. The company disclosed earlier this week that its licensing deal with Takeda could be worth up to $3.5 billion. That pact will focus on central nervous system targets.
And PeptiDream is no stranger to these high-flying deals. The Japanese company signed another with Bayer in 2017 worth as much as $1.11 billion in milestones and royalties.
Another deal with Johnson & Johnson’s Janssen is worth up to $1.15 billion, and others have been signed with Amgen, AstraZeneca, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Novartis, Merck, Sanofi and many more.
The deals could provide a slow trickle of billions for PeptiDream over the years as they deliver on their peptide promises.
With its rapid mutations, the evolutionary process of the coronavirus makes it a moving target. And so it is with treatments for the disease, which can quickly become in vogue or obsolete.
Case in point for the former: Regeneron’s antibody cocktail REGEN-COV. Over the last few months, nearly every development in the COVID-19 antibody market has broken in favor of the treatment, a combination of casirivimab and imdevimab.
On Friday, the FDA granted emergency use authorization to REGEN-COV as a preventative measure for those who have been exposed to COVID-19 and who are at high risk to progress to a severe case because they are not fully vaccinated or are not expected to mount an adequate antibody response to vaccination.
About 3% of the U.S. population won’t respond fully to COVID-19 vaccines, including patients on chemotherapy, people with hematologic cancers and others, Regeneron says.
The FDA based its decision on a phase 3 trial, run jointly with the National Institutes of Health, which showed that REGEN-COV cut the risk of developing symptomatic COVID by 81% among healthy individuals who lived with an infected person.
The study tested the med in 1,505 people who lived with an infected person within the prior four days. Those who received an infusion of REGEN-COV showed 72% efficacy against symptomatic COVID over the first week and 93% beyond the first week.
Additionally, among those who developed symptoms, REGEN-COV recipients cleared the virus faster than those in the placebo group.
While vaccine recipients need time to build an effective antibody response, especially with a two-shot regimen, a direct injection of antibodies can offer more immediate protection.
The FDA granted REGEN-COV its initial EUA in November for patients with newly diagnosed COVID-19 who are at a high risk of progressing to severe disease. Since then, the drug Regeneron’s therapy has gained FDA nods for a subcutaneous injection and at a smaller dose.
Regeneron’s antibody trailed Eli Lilly’s antibody onto the market, but it has since proven more resilient against mutations than its rival. Another antibody drug gained FDA approval two months ago.
But sotrovimab, a joint effort from GlaxoSmithKline and Vir Biotechnology, has yet to secure a supply deal from the United States.
The powerhouse of the cell is powering a new collaboration pact between Astellas Pharma and Minovia Therapeutics, with the latter receiving $20 million upfront for the joint work on cell therapies.
The Japanese Big Pharma, no stranger to biotech deals having penned a $3.5 billion deal with PeptiDream just days ago, will also pay the U.S.-Israel biotech up to $420 million in biobucks for each cell therapy commercialized for diseases caused by mitochondrial dysfunction, under the terms of the deal revealed Friday.
Mitochondrial dysfunction occurs when the mitochondria, which produce much of a cell’s energy, don’t work as well as they should due to another disease or condition.
Many conditions can lead to secondary mitochondrial dysfunction and affect other diseases, including Alzheimer’s disease, muscular dystrophy, Lou Gehrig’s disease, diabetes and cancer.
The collab license pact focuses specifically on allogeneic mitochondrial cell therapies. Astellas will contribute cells from its genetically-engineered, induced pluripotent stem cells. Minovia meanwhile will hand forth its mitochondrial augmentation therapy platform. The biotech is currently testing the MAT platform in a phase 1 study of Pearson Syndrome, a childhood bone marrow disease.
“We, at Astellas, have positioned mitochondrial biology as one of the Primary Focuses of our research and development strategy to develop therapies for patients with unmet medical needs. One of the aspirations of this Primary Focus is to establish a mitochondrial cell therapy platform,” said Astellas Chief Financial Officer Naoki Okamura in a statement.
The goal is to treat diseases by transferring healthy mitochondria to restore patients’ tissues. This involves isolating a patient’s own cells, providing them with healthy mitochondria from a donor and then re-infusing back into the patient.
The mitochondrial biology pact with Minovia follows Astellas’ deals in the space in recent years. The Big Pharma acquired Mitobridge in late 2017 and, more recently, Nanna Therapeutics in April 2020.
The Mitobridge deal appears to have produced three clinical-stage assets for the Astellas pipeline. Mitobridge is listed as a partner on Astellas’ mitochondria biology primary focus areas in a July 30 pipeline update, with a phase 2 acute kidney injury small molecule, a phase 2 primary mitochondrial myopathy small molecule and a phase 1 Duchenne muscular dystrophy treatment hopeful.
“We share with Astellas both their passion for mitochondrial science and their commitment to patients in need of new therapies,” said Natalie Yivgi-Ohana, Ph.D., Minovia’s CEO and co-founder, in a statement.
Trials mixing a first dose of the Russian-made Sputnik V vaccine with AstraZeneca’s (AZN.L) shot revealed no serious side effects and no subsequent cases of coronavirus among volunteers, the Russian Direct Investment Fund (RDIF) said on Friday.
The trial involved 50 people and began in Azerbaijan in February, RDIF, which is responsible for marketing Sputnik V vaccine abroad, said in the statement.
Full results of the trial, including data on the immune response produced by the combination vaccine, would be published next month, RDIF said.
Both the Sputnik V and the AstraZeneca shot are viral vector vaccines that are based on a first dose followed by a booster.
Such viral vector shots use harmless modified viruses as vehicles, or vectors, to carry genetic information that helps the body build immunity against future infections.
The first dose of Sputnik V is based on adenovirus type 26, while AstraZeneca’s COVID-19 vaccine, developed together with Oxford University, uses a chimpanzee adenovirus vector.
Russia on Monday gave the green light for clinical trials combining the British shot and Sputnik V to go ahead across five Russian clinics.
Watch out, GlaxoSmithKline and Johnson & Johnson, Pfizer is steaming into the respiratory syncytial virus (RSV) vaccine race in what looks like a late break that could soon become gold.
In data dropped rather quietly during second-quarter earnings this week and to little fanfare, Pfizer said a challenge study, where vaccines were given to 62 adults under 50 who were then intentionally infected with RSV to see if the shot worked, showed “100% observed efficacy against mild to moderate symptomatic infection resulting from RSV.”
This is only a peek from the phase 2a test, which is set up to assess the immunogenicity and efficacy of the bivalent protein-based vaccine candidate, RSVpreF. But the data, which came out of nowhere, could be a boon for Pfizer and came with no serious safety worries.
More specifics and details are coming, and that’s where the real analysis will come. But Pfizer likes what it sees and plans to kickstart a global phase 3 trial in adults this September based on these data, with results expected as early as the first quarter of next year.
Analysts at SVB Leerink said: “We expect this move may surprise many investors who expected GSK and J&J to lead the adult RSV vaccine race.”
After talking with Pfizer, which Leerink said had been deliberately stealthy about the program, the pharma shared a little extra data suggesting that among 31 vaccinated individuals, mean RSV viral load did not increase within 12 days after challenge, compared to a 2-3 log fold mean increases among 31 placebo group members.
The company also disclosed to the firm a symptom score measure “that suggested that vaccinated trial participants expected fewer disease symptoms.”
Pfizer is riding high on what must be one of the most savvy/lucky biotech deals ever made, when it decided last spring to work with little-known German outfit BioNTech and its mRNA tech against COVID.
The deal will now bring in, according to Pfizer’s latest estimates, $33 billion this year alone, and that’s before boosters.
“Pfizer’s aggressive timeline appears inspired by the ‘lightspeed’ operational planning carried over from their COVID vaccine efforts,” Leerink added in its note to clients.
The RSV asset is set for 2026 sales of just $255 million, according to EvaluatePharma, though the entire market could be $7 billion a year in adults alone, according to Leerink, so there is much up for grabs.
That’s a far cry from the mega blockbuster COVID vaccine, which I’m sure Pfizer won’t mind us hoping won’t be needed in 2026.
Pfizer’s surprise data drop comes amid a setback for GSK this week, which abandoned its RSV candidate in kids, while moving forward with a maternal vaccine test to protect mothers and children during and from birth. Results are expected in 2024. Pfizer is running a similar phase 3 in pregnant women. J&J, meanwhile, has an RSV vaccine, Ad26.RSV.preF, in a phase 3.
Cold viruses often don’t get much of a look when it comes to R&D. They are annoyances, but ones that usually make us feel miserable for a few days then clear up.
But some cold viruses can hit vulnerable people much harder, leading to pneumonia and hospitalizations. These are colds caused by RSV, which, in the elderly and children under 5, can cause serious complications and sometimes prove fatal.
In younger children, it can cause bronchiolitis, or inflammation and congestion of the small airways or bronchioles of the lung. The virus is thought to cause about 3 million hospitalizations of children under 5 globally, with around 177,000 hospitalizations of older people in the U.S.
A number of Big Pharmas have attempted a vaccine, but the road has been fraught with setbacks and flops. Novavax, also in the COVID running, knows this all too well.
There is a monthly preventive shot from Swedish Orphan Biovitrum, called Synagis, which can be used against RSV in high-risk infants, but a fully protective vaccine remains elusive.
After a major lull in RSV infections across the world during last winter as masks and isolation cut rates dramatically, the great re-opening could see the virus surge back, so the need for vaccines has become paramount.
The Serum Institute of India (SII) said on Friday it had partnered with an industry body to expand the reach of COVID-19 shots in India’s countryside as it had “plenty of supply” of the AstraZeneca (AZN.L) drug.
India has reported more than 31.5 million coronavirus infections in the world, the most after the United States, with 423,217 deaths.
While some 452 million vaccine doses have been administered, the pace of inoculations in rural areas, where two-thirds of India’s 1.35 billion people live, has lagged that of urban centres.
SII, the world’s largest vaccine maker, will be working with the Confederation of Indian Industry (CII), which has set up vaccination camps in smaller towns and rural areas.
“While SII has manufactured the vaccine at scale within a short period of time, it is essential that all stakeholders work together towards carrying out the inoculations,” CEO Adar Poonawalla said about its version of the AstraZeneca drug known as Covishield.
SII stopped all vaccine exports in April to meet India’s own demand and has forecast resuming foreign shipments only towards end-2021. Its monthly Covishield production is expected to nearly double to 120 million doses from April levels. read more
“India’s post-pandemic growth critically rests on how soon the eligible population is vaccinated and economic activities can return to normal,” said CII President T.V. Narendran.
Over 70 patients completed the survey, which related to the delivery of scan results for those undergoing monitoring post-surgery after a kidney cancer diagnosis.
According to the survey, up to 85% are suffering from ‘scanxiety’, also revealing that 66% of patients are waiting at least three weeks after their scan while 20% are waiting for eight weeks or longer.
The survey also found that almost half of all respondents had little confidence that their results would be communicated with them within an ‘acceptable time span’ following the scan.
“Cancer patients are under enough pressure through living with and beyond their diagnosis. The additional stress and anxiety brought on by having to wait such an unreasonable time for a scan result on the NHS is not acceptable,” said Nick Turkentine, chief executive officer of Kidney Cancer UK
“We have to ensure there is a limit on how long a patient should have to wait for these momentous scan results and end the culture of Scanxiety.
And, in line with Jonathan Perry’s comment, we should ensure our NHS is fit for purpose, staffed correctly and rewarded fully,” he added.
Following the survey findings, the UK-based kidney cancer charity is urging NHS urology departments to consider the effect of these delays on patients’ mental health and reduce the time it takes to share scan results with them.
Marc Semigran was at sleepaway camp in Hopewell Junction, NY, when Neil Armstrong took the first steps on the moon. He packed into a social hall with a couple of hundred kids whose eyes were glued to one small, staticky, black-and-white television as Armstrong descended the ladder.
It sparked an interest in physics for Semigran whose focus later turned to medicine because he wanted to help people. That, and “I’m probably too tall, I don’t think I would have made it as an astronaut,” he quipped.
He went on to Harvard Medical School, then an internship and residency at the renowned Massachusetts General Hospital.
His cardiology work there eventually led to a gig as CMO of MyoKardia, where he stayed until the company was bought out by Bristol Myers Squibb for a whopping $13.1 billion last year. And now, he’s taking the same role at gene therapy-focused Renovacor, just as the company prepares to go public on the back of a SPAC.
“Something that really reassured me about my decision was that we will have the funds to be able to carry out the scientific mission,” he said. Upon closing its deal with the blank-check company Chardan Healthcare Acquisition 2 Corp, Renovacor is set to pocket gross proceeds of up to $116 million.
Semigran joined MyoKardia back in 2016, where he oversaw Phase I and II studies and the design of registrational studies, worked with regulators, and helped file the NDA for mavacamten — MyoKardia’s drug for obstructive cardiomyopathy. In 2019, Semigran was promoted to senior VP of medical sciences.
The FDA set a decision date of Jan. 28, 2022, for mavacamten, the same year that Renovacor is hoping to get IND clearance for REN-001, its gene therapy for BAG3-associated dilated cardiomyopathy (DCM).
“It would be great,” he said, if one of his drugs gets approved in the same year that another enters the clinic.
“So many of our therapies for heart failure really address manifestations of heart failure, rather than the primary underlying problem,” Semigran said. “Differentiating by cause is really what we need to do with heart failure… Both REN-001 and mavacamten address very different segments of the heart failure population, and we need them both.”
BAG3 protein is expressed predominantly in the heart, where it plays a role in the maintenance of sarcomeres, normalization of protein quality control, inhibition of programmed cell death, and responsiveness to adrenergic signals.
BAG3 DCM has an average onset at 38 years old, and a survival rate of less than 50% five years after discovery.
“Certainly, our first and primary target is to address the patients with the genetic mutations in BAG3. But as we learn more from treating these patients, there may be the possibility of expansion to other heart failure patients,” Semigran said, or even other organs.
Mindset Pharma Inc. (CSE: MSET) (FSE: 9DF) (OTCQB: MSSTF) (“Mindset” or the “Company”), a drug discovery and development company focused on creating optimized and patentable next-generation psychedelic medicines to treat neurological and psychiatric disorders with unmet medical needs,
today announced that it has selected its lead clinical candidate, MSP-1014, a differentiated psilocybin-based compound from its Family 1 of novel, patent-pending psychedelic compounds to move forward into current good manufacturing practice (cGMP) compliant manufacturing and investigational new drug (IND)-enabling studies.
“The selection of Mindset’s first lead candidate, MSP-1014, is a significant milestone in our clinical journey. We are excited to enter the final preclinical development step prior to commencing first-in-human clinical trials.
Our next generation psychedelic compounds represent the flourishing evolution of therapeutics to effectively address neurological and neuropsychiatric disorders,” said James Lanthier, CEO of Mindset.
“MSP-1014 demonstrates superior preclinical characteristics in head-to-head comparison with psilocybin and its active metabolite psilocin, including increased safety and efficacy, which we believe will potentially result in lower clinical doses thereby indirectly decreasing safety concerns, and manufacturing advantages. MSP-1014 has the potential to be a safer, more efficacious analog to psilocybin, with reduced potential side effects.
Given its chemical profile, we anticipate that MSP-1014 will have the potential to treat mood disorders, including major depressive disorder, substance misuse disorders and end-of-life angst associated with terminal illnesses, including cancer. Therefore, we believe that MSP-1014 has the potential to be a first-in-class psychedelic drug candidate.
With our first lead candidate selected, Mindset continues to steadily advance its position in developing innovative next-generation psychedelic therapeutics that couple the life-changing potential of psychedelic drugs with novel and patentable new chemical entities that are more predictable, convenient, and safer than first-generation psychedelics. tert-butyl N-[(2R)-1-hydroxy-4-methylpentan-2-yl]carbamate suppliers Wavelength, a more than 30-year-old firm based in Petah Tikva, launched its API CDMO business in early 2020 and currently sports more than 250 customers from 50 countries. N-Boc-(2S,3S)-(-)-2-Amino-3-methyl-1-pentanol vendor & factory.
Neuroscience drug development is hindered by the challenge of getting therapeutics into the brain and other parts of the nervous system, especially for large molecule treatments that struggle to cross the blood-brain barrier, Sheikh said.
“Large molecule therapeutics are the latest treatments to be developed for neurodegenerative diseases and technologies that improve their ability to cross the blood-brain barrier will help to maximize their benefit,” Sheikh said.
In steps PeptiDream with its TfR1 binding technology, which Sheikh said has the potential to address the blood-brain barrier penetration issue.
Sheikh declined to disclose specific targets of the pact but said the two companies have “several different programs” that they hope to bring into the clinic.
The Japanese Big Pharma is betting big on neuroscience after offloading seven programs in a deal worth up to $2 billion last June with Neurocrine Biosciences.
Takeda handed over two phase 2 assets as part of that pact, saying goodbye to candidates across schizophrenia, treatment-resistant depression and anhedonia, or the inability to feel pleasure.
Takeda’s neuroscience pipeline is farther behind than areas it’s more familiar with, including oncology, gastroenterology and rare genetics and hematology.
The company did reach a milestone in the space Wednesday, though, with a breakthrough therapy designation from the FDA for its phase 2 oral orexin agonist being studied in the chronic neurological disorder narcolepsy type 1.
The Big Pharma expects its first drug approval in neuroscience to be in fiscal year 2023, with a potential first-in-class therapy for Dravet Syndrome and Lennox-Gastaut Syndrome.
In March, Takeda regained the rights to that program from Ovid Therapeutics for a $196 million upfront payment and up to $856 million total. At the time, Takeda said it would initiate phase 3 studies in children and adults with the epilepsy syndromes in the second quarter of this year.
Also that month, Takeda said it would potentially pay more than $500 million for a five-program collaboration with BridGene, including hard-to-drug neurodegenerative disease targets.
Antibodies triggered by Sinovac Biotech’s (SVA.O) COVID-19 vaccine declined below a key threshold from around six months after a second dose for most recipients, but a third shot had a strong booster effect, a lab study showed.
Chinese researchers reported the findings from a study of blood samples from healthy adults aged between 18-59 in a paper published on Sunday, which has not been peer reviewed. https://bit.ly/3zGsxQt
Among participants who received two doses, two or four weeks apart, only 16.9% and 35.2% respectively still had neutralising antibodies above what researchers regard as a detectable threshold level six months after the second shot, the paper said.
Those readings were based on data from two cohorts involving more than 50 participants each, while the study gave third doses of the vaccine or placebo to a total of 540 participants.
Researchers said it was unclear how the decrease in antibodies would affect the shot’s effectiveness, since scientists have yet to figure out precisely the threshold of antibody levels for a vaccine to be able to prevent the disease.
Apart from durable antibodies, other components in humans immune systems such as T cells and B cell memory elicited by the vaccine may also contribute to protection, researchers involved in the study said, although the study did not provide data on those factors.
“In the short-to-medium term, ensuring more people complete the current two-dose schedule of CoronaVac (Sinovac’s vaccine) should be the priority,” the paper said.
Participants in some cohorts who received a third dose of the Sinovac shot about six months after the second showed around a 3-5 fold increase in antibody levels after a further 28 days, compared with the levels seen four weeks after the second shot.
As of end-June, Sinovac has delivered more than 1 billion doses of the vaccine, a major vaccination tool in China, Brazil, Indonesia and Chile.
Other COVID-19 vaccine makers are seeking to get authorisation for booster shots. Pfizer (PFE.N) and partner BioNTech (22UAy.DE) said earlier this month they had planned to ask U.S. and European regulators within weeks to authorise a booster dose of its COVID-19 vaccine, based on evidence of greater risk of infection six months after inoculation and the spread of the highly contagious Delta variant. read more
Siti Nadia Tarmizi, a senior health ministry official in Indonesia, told Reuters on Tuesday that clinical data from the country showed the reduced antibody level is still enough to provide protection, without providing details. read more
She said the immunisation advisory board recommends a booster vaccination 12 months after the second dose.
Indonesia has started giving booster shots produced by Moderna (MRNA.O) to medical workers this month, including those who had received Sinovac doses.
Philippines’ Health Secretary Francisco Duque said on Tuesday there is no recommendation yet from the country’s vaccine expert panel on giving a booster shot but experts are discussing the possibility.
Turkey has already started offering a third dose from either Sinovac or Pfizer to some people who have got Sinovac shots.
The research paper on Sinovac’s vaccine said that the study did not test the antibodies’ effect against more transmissible variants, and that further research was needed to assess antibody duration after a third shot.
The study was conducted by researchers at disease control authorities in Jiangsu province, Sinovac, and other Chinese institutions.
Drug manufacturer Emergent BioSolutions Inc (EBS.N) said on Friday it has received inquiries and subpoenas from a swathe of U.S. authorities related to its abilities to manufacture COVID-19 bulk drug substance.
Emergent came under regulatory scrutiny after an error led to millions of vaccine doses being ruined at its manufacturing facility in Baltimore, which was producing bulk drug substance for Johnson & Johnson’s (JNJ.N) COVID-19 vaccine.
The U.S. Food and Drug Administration (FDA) in April halted operations at the plant after it discovered that ingredients from AstraZeneca’s (AZN.L) COVID-19 vaccine, also being produced there at that time, contaminated a batch of J&J’s vaccines.
Emergent in a filing on Friday said it has received “preliminary inquiries and subpoenas to produce documents” from the lawmakers, the Department of Justice, the Securities and Exchange Commission, the Financial Industry Regulatory Authority, and Maryland and New York Attorneys General.
The Department of Justice declined to comment on the matter.
Emergent said it was producing and has produced documents as required in response and will continue to cooperate with the government inquiries.
An FDA inspection in April turned up a long list of sanitary problems and bad manufacturing practices at the facility, including failure to train personnel to avoid cross contamination of COVID-19 vaccines from J&J and AstraZeneca. read more
Emergent said on Wednesday production at the Baltimore facility will now resume, following remedial actions, additional reviews and collaboration with the FDA and its manufacturing partners. read more
An FDA spokesperson said on Thursday the facility has not yet received any authorization for a COVID-19 vaccine, but may produce vaccine substance in advance of future inspections by the health regulator.
BeiGene’s Brukinsa is already approved in the small indication of mantle cell lymphoma. With a new trial win, the drug is on track to open up a much larger market.
Brukinsa outperformed the combination of bendamustine and rituximab at staving off cancer progression or death in newly diagnosed patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL), BeiGene said Thursday.
Although detailed data from the phase 3 Sequoia trial remain under wraps, BeiGene characterized Brukinsa’s advantage as “highly statistically significant.” The win was for patients whose tumor didn’t have a resistance risk factor called del(17p).
The latest win adds onto Brukinsa’s recent success in previously treated CLL/SLL and puts it on a collision course with the other two BTK inhibitors in the key CLL battleground.
In a head-to-head phase 3 trial dubbed Alpine, Brukinsa showed it could trigger a significantly better response than AbbVie and Johnson & Johnson’s market-leading Imbruvica in patients with relapsed or refractory CLL/SLL.
Plus, in an important safety win, patients who received BeiGene’s drug were less likely to develop atrial fibrillation, or irregular heartbeat.
Now, with pivotal data available from two trials, BeiGene could seek FDA nods for both front-line and second-line CLL simultaneously.
When BeiGene unveiled the second-line win at the European Hematology Association’s virtual congress last month, Jane Huang, M.D., chief medical officer of hematology, told Fierce Pharma that “there may be opportunities” to file both sets of data in one FDA application.
The new data could support an approval and a favorable label, “thus positioning Brukinsa as a key BTK inhibitor in the CLL space, particularly if the asset continues to demonstrate differentiation that could translate into a commercial advantage,” SVB Leerink analyst Andrew Berens wrote in a Friday note.
As the first BTK inhibitor to hit the scene, Imbruvica owns the lion’s share of the market. In the first half of 2021, J&J collected $2.24 billion from Imbruvica global sales, while AbbVie reported $2.65 billion, including $551 million from profit sharing with J&J.
Meanwhile, AstraZeneca’s Calquence is catching up quickly with a first-half haul of $490 million thanks to 150% year-over-year growth. The majority, $445 million, came from the U.S.
Calquence’s growth suggests that Imbruvica’s first-mover advantage in CLL isn’t insurmountable, Berens said, adding that Brukinsa seems to have an even better profile than Calquence.
Still, Brukinsa is BeiGene’s first commercial drug in the U.S. Facing those Big Pharma companies with entrenched oncology presence won’t be cakewalk for the Chinese company.
Brukinsa’s first-quarter sales came in at $22.1 million, mostly from China, where the drug has been allowed for relapsed or refractory CLL/SLL since last June.
Apparently, a licensing agreement with Takeda worth a potential $3.5 billion wasn’t enough for PeptiDream this week. The biopharmaceutical company, which has a peptide-based drug discovery platform, has now inked a similar deal with Alnylam worth a potential $2.2 billion.
Alnylam, which focuses on RNAi therapeutics, has tapped PeptiDream to develop peptide-small interfering RNA (siRNA) conjugates that will help deliver RNAi therapeutics to tissues outside the liver.
PeptiDream will receive an undisclosed upfront payment from Alnylam, R&D funding over the term of the collaboration and may be eligible to receive up to $2.2 billion in biobucks.
Additional royalty payments are part of the deal for any Alnylam products that result from the collaboration as well.That’s $5.8 billion in promise for just one week, though biotech veterans will know that the chances of every cent of that being picked up remains historically low, and comes with many, many caveats in a sector built on high-risk, high-reward.
Still, that’s a great week for PeptiDream, and further validates its platform.
“Having solved delivery of RNAi therapeutics to the liver and made substantial preclinical progress on in vivo delivery to the central nervous system, eye and lung, we are now adding to our suite of delivery technologies that have the potential to enable efficient delivery of siRNA to even broader tissue types throughout the human body,” said Alnylam’s Chief Scientific Officer Kevin Fitzgerald, Ph.D.
The collaboration will see Alnylam choose receptors for PeptiDream’s discovery platform, which will then select peptides to match.
Alnylam will then generate siRNA conjugates and perform studies to select the final peptides to move forward. The companies could develop multiple treatments from the partnership.
The companies did not disclose the exact diseases they will be working on, beyond the aim to deliver treatments to tissues outside the liver. Alnylam has four strategic therapeutic areas: genetic medicines, cardio-metabolic diseases, infectious diseases and central nervous system/ocular diseases.
This is the latest collaboration for PeptiDream that could be worth multiple billions of dollars. The company disclosed earlier this week that its licensing deal with Takeda could be worth up to $3.5 billion. That pact will focus on central nervous system targets.
And PeptiDream is no stranger to these high-flying deals. The Japanese company signed another with Bayer in 2017 worth as much as $1.11 billion in milestones and royalties.
Another deal with Johnson & Johnson’s Janssen is worth up to $1.15 billion, and others have been signed with Amgen, AstraZeneca, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Novartis, Merck, Sanofi and many more.
The deals could provide a slow trickle of billions for PeptiDream over the years as they deliver on their peptide promises.
With its rapid mutations, the evolutionary process of the coronavirus makes it a moving target. And so it is with treatments for the disease, which can quickly become in vogue or obsolete.
Case in point for the former: Regeneron’s antibody cocktail REGEN-COV. Over the last few months, nearly every development in the COVID-19 antibody market has broken in favor of the treatment, a combination of casirivimab and imdevimab.
On Friday, the FDA granted emergency use authorization to REGEN-COV as a preventative measure for those who have been exposed to COVID-19 and who are at high risk to progress to a severe case because they are not fully vaccinated or are not expected to mount an adequate antibody response to vaccination.
About 3% of the U.S. population won’t respond fully to COVID-19 vaccines, including patients on chemotherapy, people with hematologic cancers and others, Regeneron says.
The FDA based its decision on a phase 3 trial, run jointly with the National Institutes of Health, which showed that REGEN-COV cut the risk of developing symptomatic COVID by 81% among healthy individuals who lived with an infected person.
The study tested the med in 1,505 people who lived with an infected person within the prior four days. Those who received an infusion of REGEN-COV showed 72% efficacy against symptomatic COVID over the first week and 93% beyond the first week.
Additionally, among those who developed symptoms, REGEN-COV recipients cleared the virus faster than those in the placebo group.
While vaccine recipients need time to build an effective antibody response, especially with a two-shot regimen, a direct injection of antibodies can offer more immediate protection.
The FDA granted REGEN-COV its initial EUA in November for patients with newly diagnosed COVID-19 who are at a high risk of progressing to severe disease. Since then, the drug Regeneron’s therapy has gained FDA nods for a subcutaneous injection and at a smaller dose.
Regeneron’s antibody trailed Eli Lilly’s antibody onto the market, but it has since proven more resilient against mutations than its rival. Another antibody drug gained FDA approval two months ago.
But sotrovimab, a joint effort from GlaxoSmithKline and Vir Biotechnology, has yet to secure a supply deal from the United States.
The powerhouse of the cell is powering a new collaboration pact between Astellas Pharma and Minovia Therapeutics, with the latter receiving $20 million upfront for the joint work on cell therapies.
The Japanese Big Pharma, no stranger to biotech deals having penned a $3.5 billion deal with PeptiDream just days ago, will also pay the U.S.-Israel biotech up to $420 million in biobucks for each cell therapy commercialized for diseases caused by mitochondrial dysfunction, under the terms of the deal revealed Friday.
Mitochondrial dysfunction occurs when the mitochondria, which produce much of a cell’s energy, don’t work as well as they should due to another disease or condition.
Many conditions can lead to secondary mitochondrial dysfunction and affect other diseases, including Alzheimer’s disease, muscular dystrophy, Lou Gehrig’s disease, diabetes and cancer.
The collab license pact focuses specifically on allogeneic mitochondrial cell therapies. Astellas will contribute cells from its genetically-engineered, induced pluripotent stem cells. Minovia meanwhile will hand forth its mitochondrial augmentation therapy platform. The biotech is currently testing the MAT platform in a phase 1 study of Pearson Syndrome, a childhood bone marrow disease.
“We, at Astellas, have positioned mitochondrial biology as one of the Primary Focuses of our research and development strategy to develop therapies for patients with unmet medical needs. One of the aspirations of this Primary Focus is to establish a mitochondrial cell therapy platform,” said Astellas Chief Financial Officer Naoki Okamura in a statement.
The goal is to treat diseases by transferring healthy mitochondria to restore patients’ tissues. This involves isolating a patient’s own cells, providing them with healthy mitochondria from a donor and then re-infusing back into the patient.
The mitochondrial biology pact with Minovia follows Astellas’ deals in the space in recent years. The Big Pharma acquired Mitobridge in late 2017 and, more recently, Nanna Therapeutics in April 2020.
The Mitobridge deal appears to have produced three clinical-stage assets for the Astellas pipeline. Mitobridge is listed as a partner on Astellas’ mitochondria biology primary focus areas in a July 30 pipeline update, with a phase 2 acute kidney injury small molecule, a phase 2 primary mitochondrial myopathy small molecule and a phase 1 Duchenne muscular dystrophy treatment hopeful.
“We share with Astellas both their passion for mitochondrial science and their commitment to patients in need of new therapies,” said Natalie Yivgi-Ohana, Ph.D., Minovia’s CEO and co-founder, in a statement.
Trials mixing a first dose of the Russian-made Sputnik V vaccine with AstraZeneca’s (AZN.L) shot revealed no serious side effects and no subsequent cases of coronavirus among volunteers, the Russian Direct Investment Fund (RDIF) said on Friday.
The trial involved 50 people and began in Azerbaijan in February, RDIF, which is responsible for marketing Sputnik V vaccine abroad, said in the statement.
Full results of the trial, including data on the immune response produced by the combination vaccine, would be published next month, RDIF said.
Both the Sputnik V and the AstraZeneca shot are viral vector vaccines that are based on a first dose followed by a booster.
Such viral vector shots use harmless modified viruses as vehicles, or vectors, to carry genetic information that helps the body build immunity against future infections.
The first dose of Sputnik V is based on adenovirus type 26, while AstraZeneca’s COVID-19 vaccine, developed together with Oxford University, uses a chimpanzee adenovirus vector.
Russia on Monday gave the green light for clinical trials combining the British shot and Sputnik V to go ahead across five Russian clinics.
Watch out, GlaxoSmithKline and Johnson & Johnson, Pfizer is steaming into the respiratory syncytial virus (RSV) vaccine race in what looks like a late break that could soon become gold.
In data dropped rather quietly during second-quarter earnings this week and to little fanfare, Pfizer said a challenge study, where vaccines were given to 62 adults under 50 who were then intentionally infected with RSV to see if the shot worked, showed “100% observed efficacy against mild to moderate symptomatic infection resulting from RSV.”
This is only a peek from the phase 2a test, which is set up to assess the immunogenicity and efficacy of the bivalent protein-based vaccine candidate, RSVpreF. But the data, which came out of nowhere, could be a boon for Pfizer and came with no serious safety worries.
More specifics and details are coming, and that’s where the real analysis will come. But Pfizer likes what it sees and plans to kickstart a global phase 3 trial in adults this September based on these data, with results expected as early as the first quarter of next year.
Analysts at SVB Leerink said: “We expect this move may surprise many investors who expected GSK and J&J to lead the adult RSV vaccine race.”
After talking with Pfizer, which Leerink said had been deliberately stealthy about the program, the pharma shared a little extra data suggesting that among 31 vaccinated individuals, mean RSV viral load did not increase within 12 days after challenge, compared to a 2-3 log fold mean increases among 31 placebo group members.
The company also disclosed to the firm a symptom score measure “that suggested that vaccinated trial participants expected fewer disease symptoms.”
Pfizer is riding high on what must be one of the most savvy/lucky biotech deals ever made, when it decided last spring to work with little-known German outfit BioNTech and its mRNA tech against COVID.
The deal will now bring in, according to Pfizer’s latest estimates, $33 billion this year alone, and that’s before boosters.
“Pfizer’s aggressive timeline appears inspired by the ‘lightspeed’ operational planning carried over from their COVID vaccine efforts,” Leerink added in its note to clients.
The RSV asset is set for 2026 sales of just $255 million, according to EvaluatePharma, though the entire market could be $7 billion a year in adults alone, according to Leerink, so there is much up for grabs.
That’s a far cry from the mega blockbuster COVID vaccine, which I’m sure Pfizer won’t mind us hoping won’t be needed in 2026.
Pfizer’s surprise data drop comes amid a setback for GSK this week, which abandoned its RSV candidate in kids, while moving forward with a maternal vaccine test to protect mothers and children during and from birth. Results are expected in 2024. Pfizer is running a similar phase 3 in pregnant women. J&J, meanwhile, has an RSV vaccine, Ad26.RSV.preF, in a phase 3.
Cold viruses often don’t get much of a look when it comes to R&D. They are annoyances, but ones that usually make us feel miserable for a few days then clear up.
But some cold viruses can hit vulnerable people much harder, leading to pneumonia and hospitalizations. These are colds caused by RSV, which, in the elderly and children under 5, can cause serious complications and sometimes prove fatal.
In younger children, it can cause bronchiolitis, or inflammation and congestion of the small airways or bronchioles of the lung. The virus is thought to cause about 3 million hospitalizations of children under 5 globally, with around 177,000 hospitalizations of older people in the U.S.
A number of Big Pharmas have attempted a vaccine, but the road has been fraught with setbacks and flops. Novavax, also in the COVID running, knows this all too well.
There is a monthly preventive shot from Swedish Orphan Biovitrum, called Synagis, which can be used against RSV in high-risk infants, but a fully protective vaccine remains elusive.
After a major lull in RSV infections across the world during last winter as masks and isolation cut rates dramatically, the great re-opening could see the virus surge back, so the need for vaccines has become paramount.
The Serum Institute of India (SII) said on Friday it had partnered with an industry body to expand the reach of COVID-19 shots in India’s countryside as it had “plenty of supply” of the AstraZeneca (AZN.L) drug.
India has reported more than 31.5 million coronavirus infections in the world, the most after the United States, with 423,217 deaths.
While some 452 million vaccine doses have been administered, the pace of inoculations in rural areas, where two-thirds of India’s 1.35 billion people live, has lagged that of urban centres.
SII, the world’s largest vaccine maker, will be working with the Confederation of Indian Industry (CII), which has set up vaccination camps in smaller towns and rural areas.
“While SII has manufactured the vaccine at scale within a short period of time, it is essential that all stakeholders work together towards carrying out the inoculations,” CEO Adar Poonawalla said about its version of the AstraZeneca drug known as Covishield.
SII stopped all vaccine exports in April to meet India’s own demand and has forecast resuming foreign shipments only towards end-2021. Its monthly Covishield production is expected to nearly double to 120 million doses from April levels. read more
“India’s post-pandemic growth critically rests on how soon the eligible population is vaccinated and economic activities can return to normal,” said CII President T.V. Narendran.
Over 70 patients completed the survey, which related to the delivery of scan results for those undergoing monitoring post-surgery after a kidney cancer diagnosis.
According to the survey, up to 85% are suffering from ‘scanxiety’, also revealing that 66% of patients are waiting at least three weeks after their scan while 20% are waiting for eight weeks or longer.
The survey also found that almost half of all respondents had little confidence that their results would be communicated with them within an ‘acceptable time span’ following the scan.
“Cancer patients are under enough pressure through living with and beyond their diagnosis. The additional stress and anxiety brought on by having to wait such an unreasonable time for a scan result on the NHS is not acceptable,” said Nick Turkentine, chief executive officer of Kidney Cancer UK
“We have to ensure there is a limit on how long a patient should have to wait for these momentous scan results and end the culture of Scanxiety.
And, in line with Jonathan Perry’s comment, we should ensure our NHS is fit for purpose, staffed correctly and rewarded fully,” he added.
Following the survey findings, the UK-based kidney cancer charity is urging NHS urology departments to consider the effect of these delays on patients’ mental health and reduce the time it takes to share scan results with them.
Marc Semigran was at sleepaway camp in Hopewell Junction, NY, when Neil Armstrong took the first steps on the moon. He packed into a social hall with a couple of hundred kids whose eyes were glued to one small, staticky, black-and-white television as Armstrong descended the ladder.
It sparked an interest in physics for Semigran whose focus later turned to medicine because he wanted to help people. That, and “I’m probably too tall, I don’t think I would have made it as an astronaut,” he quipped.
He went on to Harvard Medical School, then an internship and residency at the renowned Massachusetts General Hospital.
His cardiology work there eventually led to a gig as CMO of MyoKardia, where he stayed until the company was bought out by Bristol Myers Squibb for a whopping $13.1 billion last year. And now, he’s taking the same role at gene therapy-focused Renovacor, just as the company prepares to go public on the back of a SPAC.
“Something that really reassured me about my decision was that we will have the funds to be able to carry out the scientific mission,” he said. Upon closing its deal with the blank-check company Chardan Healthcare Acquisition 2 Corp, Renovacor is set to pocket gross proceeds of up to $116 million.
Semigran joined MyoKardia back in 2016, where he oversaw Phase I and II studies and the design of registrational studies, worked with regulators, and helped file the NDA for mavacamten — MyoKardia’s drug for obstructive cardiomyopathy. In 2019, Semigran was promoted to senior VP of medical sciences.
The FDA set a decision date of Jan. 28, 2022, for mavacamten, the same year that Renovacor is hoping to get IND clearance for REN-001, its gene therapy for BAG3-associated dilated cardiomyopathy (DCM).
“It would be great,” he said, if one of his drugs gets approved in the same year that another enters the clinic.
“So many of our therapies for heart failure really address manifestations of heart failure, rather than the primary underlying problem,” Semigran said. “Differentiating by cause is really what we need to do with heart failure… Both REN-001 and mavacamten address very different segments of the heart failure population, and we need them both.”
BAG3 protein is expressed predominantly in the heart, where it plays a role in the maintenance of sarcomeres, normalization of protein quality control, inhibition of programmed cell death, and responsiveness to adrenergic signals.
BAG3 DCM has an average onset at 38 years old, and a survival rate of less than 50% five years after discovery.
“Certainly, our first and primary target is to address the patients with the genetic mutations in BAG3. But as we learn more from treating these patients, there may be the possibility of expansion to other heart failure patients,” Semigran said, or even other organs.
Mindset Pharma Inc. (CSE: MSET) (FSE: 9DF) (OTCQB: MSSTF) (“Mindset” or the “Company”), a drug discovery and development company focused on creating optimized and patentable next-generation psychedelic medicines to treat neurological and psychiatric disorders with unmet medical needs,
today announced that it has selected its lead clinical candidate, MSP-1014, a differentiated psilocybin-based compound from its Family 1 of novel, patent-pending psychedelic compounds to move forward into current good manufacturing practice (cGMP) compliant manufacturing and investigational new drug (IND)-enabling studies.
“The selection of Mindset’s first lead candidate, MSP-1014, is a significant milestone in our clinical journey. We are excited to enter the final preclinical development step prior to commencing first-in-human clinical trials.
Our next generation psychedelic compounds represent the flourishing evolution of therapeutics to effectively address neurological and neuropsychiatric disorders,” said James Lanthier, CEO of Mindset.
“MSP-1014 demonstrates superior preclinical characteristics in head-to-head comparison with psilocybin and its active metabolite psilocin, including increased safety and efficacy, which we believe will potentially result in lower clinical doses thereby indirectly decreasing safety concerns, and manufacturing advantages. MSP-1014 has the potential to be a safer, more efficacious analog to psilocybin, with reduced potential side effects.
Given its chemical profile, we anticipate that MSP-1014 will have the potential to treat mood disorders, including major depressive disorder, substance misuse disorders and end-of-life angst associated with terminal illnesses, including cancer. Therefore, we believe that MSP-1014 has the potential to be a first-in-class psychedelic drug candidate.
With our first lead candidate selected, Mindset continues to steadily advance its position in developing innovative next-generation psychedelic therapeutics that couple the life-changing potential of psychedelic drugs with novel and patentable new chemical entities that are more predictable, convenient, and safer than first-generation psychedelics. tert-butyl N-[(2R)-1-hydroxy-4-methylpentan-2-yl]carbamate suppliers Wavelength, a more than 30-year-old firm based in Petah Tikva, launched its API CDMO business in early 2020 and currently sports more than 250 customers from 50 countries. N-Boc-(2S,3S)-(-)-2-Amino-3-methyl-1-pentanol vendor & factory.
